On June 30, the research team headed by GUO Guoji, HAN Xiaoping and WANG Jingjing at the Zhejiang University School of Medicine published an open-access article titled “An analytical framework for decoding cell type-specific genetic variation of gene regulation” in the journal Nature Communications.
Since the completion of Human Genome Project two decades ago, substantial progress has been made in the field of genomics. However, the vast majority of human genome sequences, consisting of 3 billion base pairs, are non-coding regions, and the human understanding of their potential functions is only the tip of the iceberg. In the past decade, a great deal of work has been committed to exploring the regulatory mechanisms of genetic expression on a genome-wide scale. Despite the fact that expression quantitative trait loci (eQTLs) and eGenes have considerably facilitated the functional interpretation of the genome-wide association study (GWAS) findings, a deeper understanding of the underlying biological mechanisms has been hindered by heterogeneous cellular compositions in bulk tissues.
GUO Guoji et al. made two major breakthroughs in methodology in their study. Firstly, they proposed a novel approach to inferring cell type-dependent eQTLs based on single-cell expression profiles. This approach aims to uncover more associations between non-coding mutations and genes concealed in traditional analyses. Secondly, they improved the existing deep learning model and developed an effective method for building a cell type-specific predictive model based on sing-cell expression profiles. By simulating base substitutions in DNA sequences, the model is able to predict the transcriptional-regulation-disrupting effect of mutations in each cell type. Finally, they integrated these methods into a framework called Huatuo and developed it into a user-friendly tool for researchers. This framework facilitates the exploration of cell landscapes and genome-wide genetic variations in cell type-specific regulation using scRNA-seq data from a small cohort of individuals.

To validate the feasibility of this analytical approach, the researchers systematically evaluated the predictive performance of the Huatuo model. They found that the Huatuo model could accurately predict the level of gene expression on the strength of DNA sequences. Across 357 cell clusters derived from 20 different tissues, the model achieved a median Pearson’s correlation coefficient (PCC) of 0.763 between the predicted and actually observed gene expression levels. Some cell clusters of the kidneys, stomachs and transverse colons even obtained a PCC higher than 0.80. In addition, to assess the plausibility of the prediction results, they also tested whether the model could reproduce eQTL results solely based on DNA sequences. Although some loci with very large transcriptional-regulation-disrupting effects may be rare due to negative selection effects, thus leading to a lack of statistical power of eQTLs, the results still showed a significant correlation between the highest absolute value of variant predictions and the maximum eQTL z-score within the same linkage disequilibrium (LD) block.
Besides, the researchers also evaluated the analytical results of cell-dependent genetic associations. By performing Bayesian co-localization on 114 GWAS datasets, they found that the cell cluster-ieQTLs computed by Huatuo revealed co-localization signals that were undetectable using standard eQTLs. They then employed a “Silver Standard” dataset to examine the colocalized results. This colocalization analysis displayed a large number of colocalized signals that were not observed with standard eQTLs, indicative of significant enrichment for the gene-trait in the “silver standard” dataset. Overall, these results demonstrate the biological plausibility of the putative cell cluster-ieQTL results predicted by Huatuo.
Using the Huatuo framework, researchers hypothesized the regulatory effects of cell-dependent eQTLs as well as all common mutations in the population in different cell clusters. They constructed a comprehensive landscape of cell type-specific genetic variations for 44 major cell types. This landscape identified a total of 13,182 cell type-specific functional regulatory variants and 6,181 associated genes that are likely to be perturbed by the putative causal regulatory variants.
Thanks to 114 GWAS datasets, the researchers comprehensively assessed the contribution of the Huatuo landscape to the heritability of various complex diseases and traits. Based on the Huatuo landscape, they estimated the enrichment of SNP-based heritability at cell type-specific regulatory loci, demonstrating its potential in uncovering disease-driving cell types. Moreover, they provided insights into the functional mechanisms at the single-base and cell-type levels for fine mapping GWAS causal mutations. To better present these results, they created a database website for Huatuo genetic variants (http://bis-zju-edu-cn-s.webvpn.zju.edu.cn:8001/huatuo/) to facilitate the application of the generated data resources in future studies.
In brief, this study addresses the bottlenecks caused by technical deficiencies and sampling difficulties, and offers a new paradigm for decoding the function of non-coding mutations and studying disease genomes. These findings will push forward the field of functional genomics, thereby laying a foundation for elucidating cellular pathways critical for disease development and achieving precise and customized medicine.
The Fourth Affiliated Hospital of Zhejiang University School of Medicine has achieved a significant milestone by performing China's first interventional operation combining a leadless pacemaker wi...
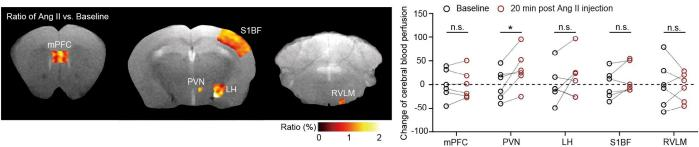
High arterial blood pressure (A.K.A. hypertension) attacks over 1/3 of adult population worldwide. As a “silent killer”, hypertension exacerbates lethal outcome of cardiovascular diseases such as he...
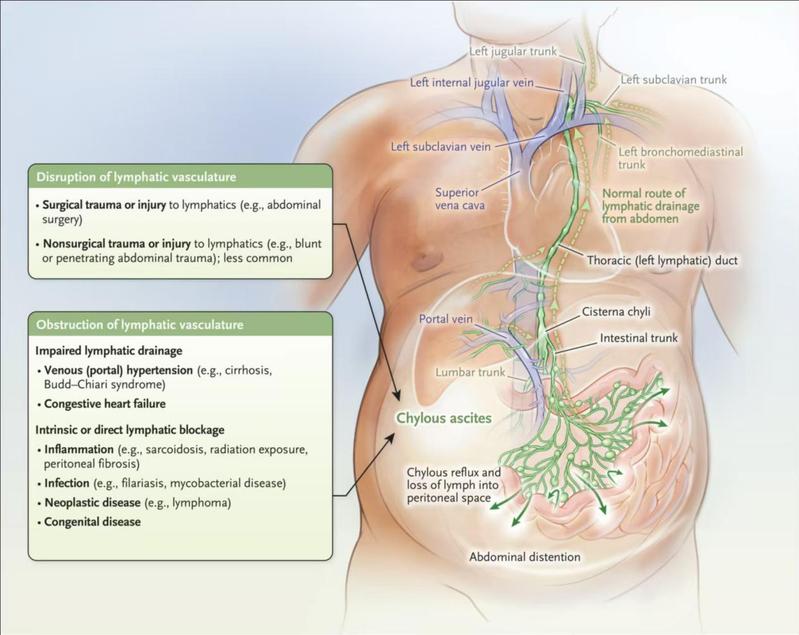
Chyle leak (CL) following pancreatectomy primarily results from iatrogenic injury to the cisterna chyli or its tributaries during pancreatic resection procedures. Having been overlooked for decades, C...